What's new in CodonCode Aligner 3.5
CodonCode Aligner version 3.5 introduced several new features and improvements like building Neighbor-Joining trees, zooming in the contig view, shortening reference sequences after alignments, masking contig bases in low coverage regions, the percentage consensus as additional consensus method, and much more.
Version 3.7.1 added the option to replace '-' characters in consensus sequences with 'n', and included a number of important bug fixes.
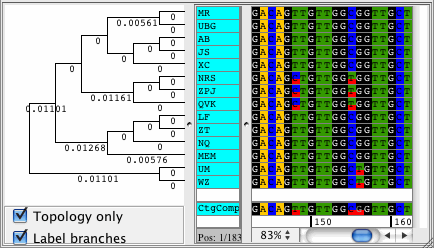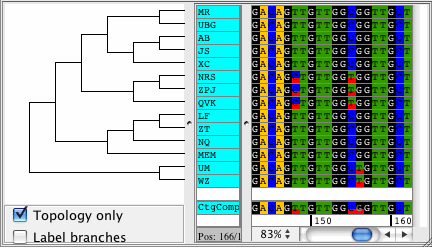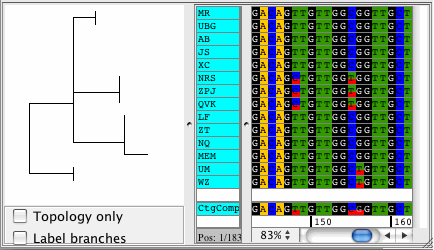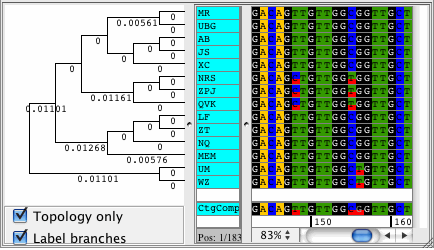
Automatically shorten reference sequence after alignmentsUncovered reference sequences can now be shortened automaticaly after an alignment in CodonCode Aligner. Choose from clipping the reference sequence to a number of uncovered bases on each side, clipping to the alignment or the exon, or not clipping at all. This new feature allows automatic clipping of very long reference sequences to the region you want to look at. For example, a 100,000 bases long genomic reference sequence can be clipped to single exon regions after an alignment. |
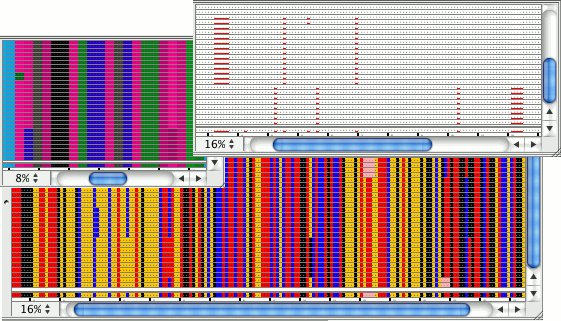
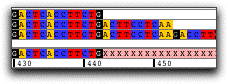
Mask low coverage regionsAutomatically mask consensus bases in regions with low coverage. If the coverage is less than what you specified, CodonCode Aligner can change the consensus base to 'N', 'X', or '-' .
In the example above, regions with lower than 3-fold coverage are indicated by using 'X' as the consensus base. |
Percentage consensusInclude only differences that occur in a specific amount of your sequences by using a percentage consensus when building your assembly. The percentage consensus complements the existing options of quality-based, majority, and inclusive consensus, or using the reference sequence as consensus. Also new is the ability to use different consensus methods for regular contigs (for example a quality-based consensus) and contigs of contigs (for example a percentage-based or inclusive consensus). |
More...
- Set base numbers: Set the base number for bases in samples and contigs. This allows you to set and view base numbers relative to (for example) cloning sites or coding sequences.
- Automatic name scheme definition for "Assemble & Align by Name": The feature "Assemble in Groups" (by name) now offers the option to automatically define a name scheme for the samples in your project. CodonCode Aligner can guess a name scheme based on the delimiters used in your sample names.
- Customize toolbars directly through popup menus: Just right-click on the toolbar in each view to customize the toolbars through a popup, adding buttons for features you use often, and removing buttons for features you do not use.
- Support for bi-directional scrolling: For users of mice that support bi-directional scrolling, like Apple's Magic Mouse, we added the option to change the scrolling axes.
- Rebuild the consensus: Use the popup menu or the menu item to rebuild the consensus; for example after opening a project with different settings.
- Speed improvements for projects with many contigs
CodonCode Aligner 3.5 has been fully tested and is compatible with Mac OS X 10.6 (Snow Leopard) and Windows 7.
Interested? Watch the movie showing the new features of CodonCode Aligner 3.5.
The new features and improvements listed above describe what is new in CodonCode Aligner version 3.5. The CodonCode Aligner version history shows a comparison of the features available in different versions. New features introduced in previous Aligner releases are described separately for CodonCode Aligner 3.0, 2.0, 1.6, 1.5, and 1.4.