
In this section, we will show how you can exclude regions from mutation finding. Examples where this can be useful are:
To exclude regions from mutation finding by Aligner, you need to add a "dontGenotype" tag to the region. You can add these tags to individual samples to exclude just the sample, or to the consensus sequence to exclude a region completely (for all samples). Let's get started:
You should now see a trace window with two traces (possibly more if you have automatic trace selection turned on). Use the vertical scroll bars on the right to increase the height of the traces so they look like this:
Look closely at the 't' that carries the pink heterozygote tag. There is also a peak in the A-lane (green) that has the same intensity. This seems like a heterozygous base until you look at the other trace, and notice that the three A's here are look exactly the same as the three A's below. The height of the first two A peaks is about the same, the third A peak is higher. If this was a real heterozygous base, you would expect the first A peak in the lower sequence to be only half the intensity of the next peak (or the peak in the upper sequence). Overall, both traces have quite a bit of random "noise" peaks - so we conclude that the red "t" peak at contig base 107 in va-1.x is just noise, not a real peak (you may not agree with this analysis, but just bear with me, ok?).
To tell Aligner to not look at this base again, we will add a "dontGenotype" tag to this location in the sample. Here is how:
You "Add tag" dialog should now look like this:
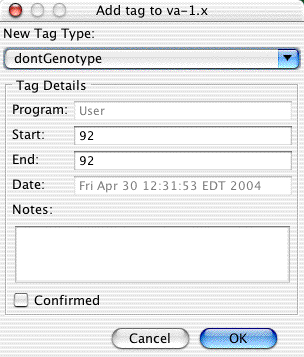
Note that the position shown is relative to the sample, not to the contig. This sample starts at base 16 in the contig, so base 92 in the sample is at base 107 in the contig.
Click "OK" to add this tag. You should see that the tag color changes from pink (for heterozygote) to light blue (for multiple tags at this base) .
Next, also add a "dontGenotype" tag to the sample "va-23.x" at this position.
Now repeat the mutation finding for this contig (if you still have the trace view in the foreground, you may need to first click on the contig view, or select the contig in the project view). This time, your results should look like this:

Note that Aligner did not find any mutations at base 107 this time, since we told it to ignore the two traces that had the "noise" peaks at this position.
You can add more than one "dontGenotype" tag to a sample. You can also add "dontGenotype" tags to the consensus sequence - this will make sure that none of the samples at this position will be analyzed. If you wanted to analyse only base 100 to 199 in a contig, you could add two "dontGenotype" tags to the consensus, one from base 1-99, and a second one from base 200 to the end of the contig.
When adding tags to the consensus, keep in mind that consensus tags will be removed by Aligner when you add additional samples to the contig (we hope to change this in future releases). If you have a reference sequence for your samples, you can add the "dontGenotype" tags to the reference sequence instead. Then, the tags on the reference sequence will be used, as long as (a) your contig was produced by alignment to the reference sequence (as opposed to assembling), and (b) you selected to use the reference sequence as the consensus in the "Consensus method" preferences.
This concludes our section about finding point mutations - for additional information, please check the online help. The next section will describe how to find heterozygous insertions and deletions in sequence traces.
Aligner Home Page - Quick Tour Start - Previous - Next: Finding Heterozygous Indels