
CodonCode Aligner lets you identify heterozygous insertions and deletions ("indels"), and analyze them. This page shows how to find heterozygous indels; how to analyze them is described on the next page.
If you still have a project open, close it (no need to save any changes). Then, open the "Hetero_indel" project in the "Example Files" folder in the "CodonCode Aligner" folder. When Aligner is done reading the project, you will see a project view with two samples in the "Unassembled Samples" folder.
Select the "Unassembled Samples" folder, and then choose "Assemble" from the "Contig" menu (while Aligner can find heterozygous indels in unassembled samples, further analysis requires that the samples are assembled into a contig, so we might as well assemble now).
The assembly will create one contig. Select the contig in the project view, then go to the "Sample" menu and choose "Find heterozygous indels". You will see a progress bar for a few seconds, and then Aligner will show a dialog that summarizes the result:
Click "OK", and then look at the result table that Aligner has opened:
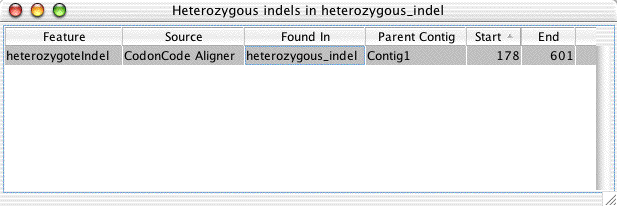 Double-click
on the line that describes the indel to open the contig view and trace view
for this sample:
Double-click
on the line that describes the indel to open the contig view and trace view
for this sample:

Aligner added a "heterozygoteIndel" tag that extends from the start of the indel to the end of the sequence. The tag is shown as a pink box under the bases. In the trace view, you can see a clean sequence up to the start of the indel tag, and many double-peaks afterwards:

This is a typical example for a heterozygous insertion or deletion. On the next page, we'll see how Aligner can help you to analyze it.
Aligner Home Page - Quick Tour Start - Previous - Next: Analyzing Heterozygous Indels