PolyPhred
PolyPhred is a program identifying heterozygous single base substitutions in assemblies of DNA sequence traces. This page gives a brief description of PolyPhred. PolyPhred is used together with the programs Phred (for base calling and peak characterization), Phrap (for assembly), and Consed or CodonCode Aligner (for viewing, editing, and annotating results).
PolyPhred was developed in Dr. Deborah Nickerson's lab at the University of Washington in Seattle, and is distributed by CodonCode under license from the University of Washington.
PolyPhred identifies putative heterozygous single base substitutions in assembled collections of DNA sequence traces. It is used together with the other programs of the Phred-Phrap-Consed package, as shown in the following diagram:

PolyPhred identifies potential heterozygous single-base substitutions by going the all bases in the Phrap-generated contigs, and examining the information about the sequence quality and peaks in each trace. For increased accuracy, PolyPhred ignores the lower quality sequences at the beginning and end of sequence traces. In the regions of sufficiently high quality, PolyPhred looks for the following characteristics that indicate a heterozygous substitution:
- A reduction in relative peak height compare to the other traces
- A secondary peak.
The following example shows a homozygous wild type sequence on top, and trace with a heterozygous point mutation below it:
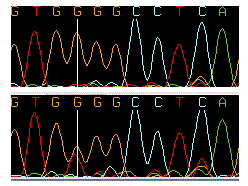
Note that the second G peak in the sequence at the bottom is only half as high as the peak in the wild type sequence, and that a second red peak indicates that this sequence is a G-T heterozygote.
PolyPhred ranks all putative point mutations it identifies on a scale of 1 to 6, with a 1 indicating highest confidence. It assigns "tags" to all the bases at this point in each sequence, indicating whether a sequence is classified a homozygous or heterozygous sequence at this position, as shown below.
PolyPhred is available for Linux, Unix, and Mac OS X, but not for Windows. For mutation detection on Windows (and OS X), CodonCode Corporation offers CodonCode Aligner. In addition to detecting heterozygous point mutations (SNPs), CodonCode Aligner also can detect and analyze heterozygous insertions and delections.
Additional information about PolyPhred can be found in the following publications:
Nickerson, D.A., Tobe, V.O., and Taylor, S.L. 1997. "PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing", Nucleic Acids Research, 25(14), pp. 2745 - 2751. [ Abstract]
Nickerson, D.A, Taylor, S.L., Weiss, K.M., Clark, A.G., Hutchinson, R.G., Stengard, J., Salomaa, V., Vartiainen, E., Boerwinkle, E., and Sing, C.F. "DNA sequence diversity in a 9.7-kb region of the human lipoprotein lipase gene." Nature Genetics, 19: 233-240, 1998. [ Abstract]
Rieder, M.J., Tobe, V.T., Taylor, S.L., and Nickerson, D.A., 1998, "Automating the identification of DNA variations using quality-based fluorescent resequencing: Analysis of the human mitochondrial genome", Nucleic Acids Res. 26: 967-973. [ Abstract]
Screen Shots
The following screen shots shows heterozygous mutations identified by PolyPhred as seen in Consed:

At base number 119, three traces are classified as heterozygous, as indicated by the pink box at the lower half of the bases, and the three other traces are homozygous C's, as indicated by the blue boxes. Clicking on the bases pops up the sequences traces; the traces for the top three sequences are shown in the next image:
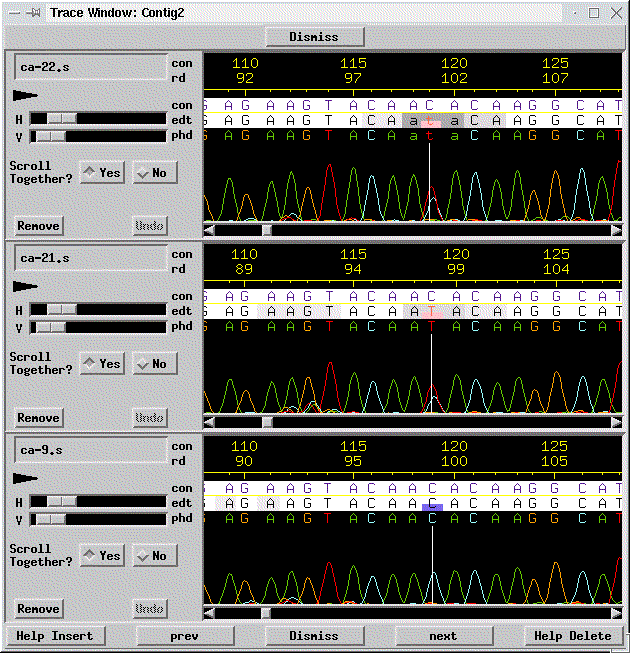
In this example, it is easy to see that the top two traces are heterozygous C-T. Often, heterozygous bases are less obvious, because the secondary peak is small or missing; PolyPhred often can correctly identify thess mutations, too, since it looks for the relative drop in intensity that can typically be seen even if the secondary peak is not obvious.
Of course, PolyPhred's performance is not perfect, and it depends on the data quality - the better the sequences are, the higher PolyPhred's accuracy will be.
Limitations
PolyPhred is a research tool, and not perfect. PolyPhred identifications contain both false positive and false negative errors - PolyPhred misses some existing mutations, and falsely flags non-mutated bases as putative substitutions. PolyPhred only identifies heterozygous single-base substitutions, not homozygous substitutions or heterozygous insertions or deletions. PolyPhred requires Phred, Phrap, and Consed, and is available only for Linux and certain UNIX operating systems (see below).
Another limitation results from PolyPhred's reliance on Phrap-generated assemblies. Phrap sometimes creates more than one contig for a given set of sequences that all cover the same region. This often happens if the sequences contain homozygous mutations - Phrap (which was originally developed for shotgun assembly, not for mutation analysis) thinks that the traces belong to two different copies of a repeat, and therfore puts them into separate contigs.
Please note: PolyPhred is intended for research use only. Polyphred has not been validated in clinical settings, and no clinical decisions should be based on PolyPhred results.
PolyPhred Availability and Prices
PolyPhred is available for Sun Solaris, Linux, and MAC OS X. PolyPhred use requires that you have a current license for Phred, Cross_match, Phrap, and Consed.
A single site license for PolyPhred costs $3,100 for standard purchase orders, and $3,000 for prepaid orders (credit cards, wire transfers, and checks are accepted for prepayment).
Academic Users
Academic users and users at non-profit organizations can obtain PolyPhred for free directly from Dr. Deborah Nickerson. For more information, please visit http://droog.mbt.washington.edu/poly_get.html.