
After finding a heterozygous indel, you will want to know the nature of the insertion or deletion. This page shows how Aligner can help you by "processing" heterozygous indels.
In the "hetero_indel" project that is still open from the previous step, select the contig, and then select "Process heterozygous indels" from the "Contig" menu. You will see a series of progress dialogs while Aligner performs the following steps:
The entire processing should be faster than reading the explanation above, so let us look at the results. In the project view, you can see that the contig "Contig1" now consists of 3 samples - the wild type sequence, the sample with the heterozygous indel, and the subtracted sequence.
Double-click on the contig to open the contig view. Select the contig view, and scroll to base 180 (the fastest way is to go to the "Go" menu and select "Base number..."). Your contig view should look like this:
Note that the new subtracted sequence, called "hetero_indel_sub", has exactly the same sequence as the wildtype, except for a one-base 'T' insertion at the start of the indel tag. To look at the traces here, double-click on the gap character (the '-') in the consensus sequence to bring up the trace view:
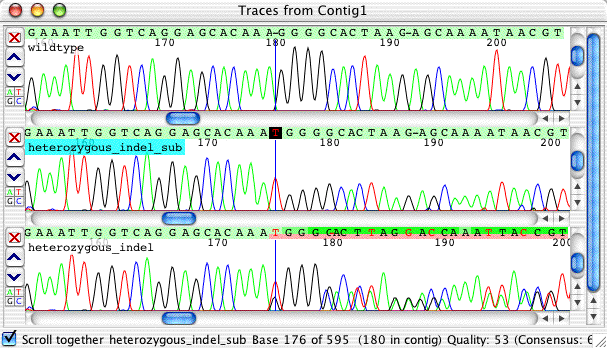
You can see that the subtracted sequence look rather nice and readable, and clearly shows the one base T insertion. You should always double-check the subtraction results by looking at the original trace, the wild type sequence, and the subtracted sequence. In this example, it is pretty easy to see that the result is correct.
One way of manually analyzing heterozygous indels is to look at only one of the four colors in a trace. The easiest color to look at is the one with the fewest peaks; in our example above, the blue C-lane is a good choice. To hide the other colors, click on the little buttons on the left of each trace that are labeled "A", "T", and "G":

Looking just at the C's, we notice that the single C in the wild type has become a double-C in the indel sequence; this indicates a one base insertion or deletion. When the peaks before the indel are aligned properly (as they are above), you can also see that the extra peak in the indel sequence is one base after the original peak - so we have a one base insertion. For comparison, the picture above also shows the result of the subtraction for this lane. For this picture, we manually deleted the gap character introduced in the wild type sequence during the alignment, so that you can "see" the subtraction: middle - top = bottom.
You can look at the other colors in isolation to find the indel. Here; it's a T, as shown clearly in the T traces:

That one extra peak before the peak double starts jumps right out, doesn't it?
The manual analysis as illustrated above can also be useful in cases where Aligner's algorithm fails, for example because of low sequence quality, additional mutations, etc. We've been able to analyze a 7-base deletion that was present in only 25% of the template material and has another mutation close to the start of the indel this way (looks like the PCR had amplified a pseudo-gene as well as the gene in this case).
Aligner's indel processing algorithm does not make any assumptions about the size of the insertion and deletion. It should work regardless of size of the indel, as long as the other allele is identical to the wild type, and a sequence without an indel, but with similar peak patterns before the indel, is available for subtraction.
Aligner Home Page - Quick Tour Start - Previous - Next: Exporting Results