What's new in CodonCode Aligner 3.0
|
|
See all amino acid translationsSimply switch between showing bases and amino acid translations for each of your samples. Choose between translation-based background colors, colored nucleotides, or quality-based background colors. Independent from the background, select to see the bases or the amino acid translation for your sequences. Quickly change the reading frame by toolbar button or menu. See and print translations in contigs, with chromatograms, or when looking at base views. |
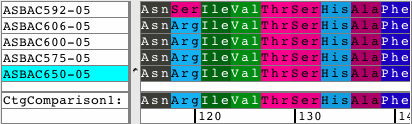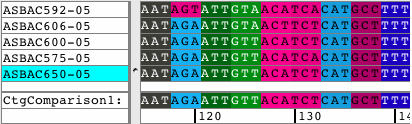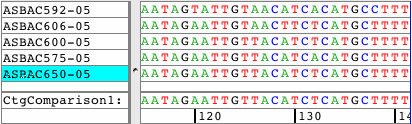
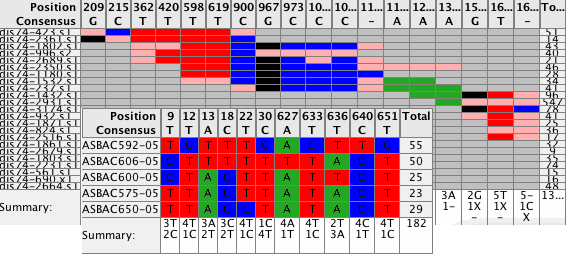
Spot the differencesEasily spot the differences in your contigs by looking at the new Difference Table. Navigate to these differences simply by clicking on any cell in the difference table. Use different filters to control what to display in your Difference Tables. Switch between compact size for overviews and a larger size for details. Export the results. |
 |
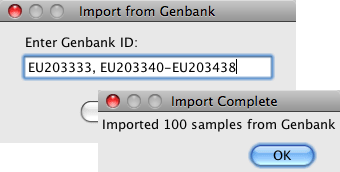
Import from GenBankImport sequences directly from GenBank by accession number. Specify multiple sequences or ranges to import hundreds of sequences into your CodonCode Aligner project.
|
Automate with scriptsUse scripts to facilitate your daily work. Many new script commands make scripting in CodonCode Aligner better than ever. Choose to run a script when starting CodonCode Aligner or access your scripts quickly through the new script menu.
Create new text sequencesEasily create new text sequences using copy and paste. CodonCode Aligner automatically parses known formats like Genbank or FASTA from the clipboard to create single or mutiple new sequences. |
Do more, fasterReduced memory use allows you to work with 3 x larger projects. Assembly and alignment has been optimized to be substantially faster when working with thousands of samples. Import 454 data from SFF files, and assemble by multiplex ID tags.
Example of substantially faster assemblies on a test data set. Speed improvements increase with project size and depth of coverage. |

More...
- Improved handling for pen tablets
- More control when exporting in FASTA format: Control the replacement of spaces or other problematic characters in sequence names. Append the number of sequences in contigs to the file. Append comments. Add gaps at the start and end of sequences in contigs, to allow import of aligned contigs in other programs.
- Automatic memory handling on Windows: CodonCode Aligner will now automatically use as much memory as is needed and available (up to 1.4 GB), without the need to edit .ini files.
- Monitor memory use in the project view: Wondering if you can add more sequences to you project? You can now monitor how much memory CodonCode Aligner uses directly in the project view.
- Remove leading and trailing contig gaps at import: Gaps are removed automatically from the start and end of sequences when importing contigs from FASTA (and similar) files.
- Contigs sorted by size: After assemblies, contigs are now sorted by the number of samples they contain before naming.
- Improved end clipping: End clipping for sequences that contain many Ns (with unreasonably high quality scores due to a KB basecalller bug) now works better.
- Paste text sequences directly into the base view
- Open read-only projects in Aligner
- Many other improvements and bug fixes
Watch the movie for a demonstration of the new features.
The new features and improvements listed above describe what is new in CodonCode Aligner version 3.0. The CodonCode Aligner version history shows a comparison of the features available in different versions. New features introduced in previous Aligner releases are described separately for CodonCode Aligner 2.0, 1.6, 1.5, and 1.4.